Einführung
Für diese Empfehlungen wurden eigene Erfahrungen sowie Erfahrungen
englischer, französischer und amerikanischer Kollegen an großen Zentren
verwertet.
Internet Fassung Sommer 2024 info(at)sichelzellkrankheit.de
Vorwort
Die Sichelzellkrankheit ist eine erbliche Hämoglobinerkrankung, die
zu rezidivierenden Gefäßverschlüssen, erhöhter Infektanfälligkeit und
chronischer hämolytischer Anämie führt. Betroffen sind Individuen aus
dem Mittelmeerraum, dem Vorderen Orient, Asien, Afrika. Die Erkrankung
kann durch die Knochenmarktransplantation, nicht aber medikamentös
geheilt werden. Inzwischen gibt es eine Gen-Therapie, die noch
experimentell ist und nur wenigen Patienten zu Verfügung stehen wird.
Durch Prophylaxe und gezielte Behandlung der vielfältigen
Organmanifestationen kann den Patienten jedoch erheblich geholfen
werden. Dieser Leitfaden soll dem betreuenden Arzt die wichtigsten
Schritte beim Management der häufigsten klinischen Probleme von
Sichelzellpatienten aufzeigen.
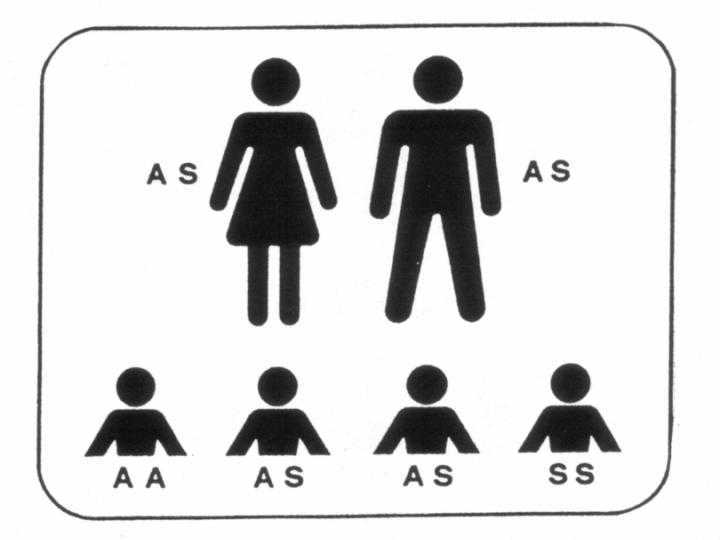
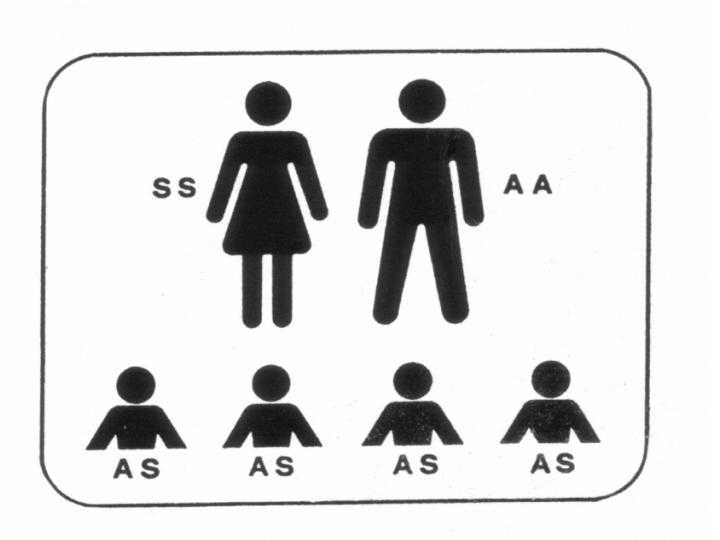
Heterozygotie für HbS (Trägerschaft)
Heterozygote Anlageträger für HbS haben durch die Trägerschaft keine
Anämie, keine Blutbild-Veränderungen, keine Schmerzkrisen oder sonstige
Krankheitsmanifestationen der Sichelzellkrankheit bis auf folgende
Ausnahmen:
Bei ca. 4% der HbS-Anlageträger kommen Episoden von
schmerzloser Makrohämat u
rie auf dem Boden von Papillennekrosen vor.
Oberhalb von 2500 - 3000 m Höhe (z.B. Bergwanderungen) können
HbS-Träger Milzinfarkte bekommen, die sich durch starke
Schmerzen im linken Oberbauch manifestieren. Flugreisen führen zu keinen
Komplikationen.
Von dem sehr seltenen medullären Nierenkarzinom
sind überdurchschnittlich häufig jüngere (10 - 40 Jahre) männliche
HbS-Träger betroffen
Schmerzen bei HbS-Trägern dürfen nicht auf
die Trägerschaft zurückgeführt werden sondern
erfordern eine Abklärung der Ursache.
Die HbS-Trägerschaft ist vor allem für die nächste Generation
wichtig. Die Partner/innen von HbS-Trägern sollten auf Trägerschaft für
HbS und ßThalassämie untersucht werden, um gegebenenfalls dem Paar
pränatale Diagnostik anbieten zu können.
Routinebetreuung
Klinische Untersuchung (Lebensalter)
| > < 6 Monate |
> alle 4 Wochen |
| > 6 Mo - 12 Mo |
> alle 2 Monate |
| > 1 - 5 Jahre |
> alle 3 Monate |
> > 5
> Jahre |
> alle 4 Monate |
| > > 10 Jahre |
> alle 6 Monate |
Routine-Ambulanzbesuche sind u.a. wichtig, um den Patienten auch in
gutem Zustand zu sehen. Dies erleichtert die Beurteilung des Patienten
in der Schmerzkrise und ermöglicht eine bessere Einschätzung der
Schmerzintensität.
Alle Patienten mit Sichelzellkrankheit sollten im Besitze eines
Ausweises sein mit genauer Angabe der Hämoglobinanomalie, dem Datum und
Ort der Diagnosestellung. Da Sichelzellpatienten oft verschiedene Ärzte
sehen, können auf diese Art überflüssige Doppelbestimmungen der
Hb-Elektrophorese vermieden werden. Ausweise für Sichelzellpatienten
können angefordert werden unter info@ist-ev.org
Labor; Spezielle Untersuchungen
- Blutbild, Retis,
Hb-Analyse, Leber-und Nierenwerte, Blutgruppe
mit
Untergruppen (Rhesus, Kell), Familienuntersuchung
und
genetische Beratung
- Vit D Spiegel |
- 1. Besuch |
| - Blutbild, Retis |
- bei jedem
Ambulanzbesuch |
- Leber-und
Nierenwerte, Urinstatus ab 5. Lebensjahr
-
TCDS 2. - 16.
Lebensjahr bei HbSS, HbSD, HbSß°Thal ,HbSOArab
Patienten |
- jährlich |
| - US Abdomem |
- jährlich ab 5.Lebensjahr |
| - Augenarzt (Fundoskopie) |
- jährl. ab 7. Lebensjahr bei HbSC Patienten,
ab 10.Lebensjahr Bei HbSS/SD/Sß°Thal |
| - Herz-Echo |
- jährlich ab 10. Lebensjahr
|
Impfungen
- DPT, Polio, HIB, MMR,
Hepatitis
- Influenza-Impfung ab
6.
Lebensmonat
- Meningokokken |
- Standard-Impfplan
- jährlich |
> Konjug.
> Pneumokokken-Impfstoff (Prevenar 13 oder 15) |
- ab 2. Lebensmonat |
| > Pneumovax |
- 2.Geburtstag (wird inzwischen in Frage gestellt)
|
Ungeimpfte Sichelzellpatienten, die vor dem 18. Lebensjahr
diagnostiziert werden, sollen zwei Prevenar 13 oder Prevenar 15 Impfungen im Abstand von
2 Monaten erhalten oder 1 Dosis Prevenar 20, gefolgt 2 Monate später von der ersten
Pneumovax-Impfung. (wird inzwischen in Frage gestellt). Der konjugierte Impfstoff löst eine wesentlich
bessere Immunantwort aus als Pneumovax. Pneumovax soll nur einmal nach 5
Jahren als Booster gegeben werden.
Kinder bis zum 18. Lebensjahr, die bereits eine Pneumovax Impfung,
aber noch nie Prevenar bekommen haben, sollen 2 x im Abstand von 2
Monaten zusätzlich Prevenar 13 geimpft werden (CDC-Empfehlung).
Penizillin - Prophylaxe (mindestens bis 5.
Lebensjahr):
| - 2 x 125 000 I.E./die |
- ab 3. Lebensmonat |
- 2 x 250 000
I.E./die |
- ab 3. Lebensjahr |
| - 2 x 500 000 I.E./die |
- ab 10.Lebensjahr |
Folsäure-Substitution (0,5-1mg/d) ist nicht
notwendig bei normaler ausgewogener Diät. Der Nutzen hoher Folsäuredosen zur evtl. Senkung des bei Sichelzellpatienten
erhöhten Homozysteinspiegel ist noch unklar und bedarf weiterer
Studien.
Medikamente
- kontraindiziert
Es gibt2 Medikamente (Ceftriaxon, G-CSF) für die es bei
Sichelzellpatienten eine strenge und eines (Cortison), bei dem es eine
relative Kontraindikation gibt:
1. Ceftriaxon (Rocephin)
- nach Ceftriaxon ist es bei Sichelzellpatienten zu schwerer, z.T.
letaler Hämolyse gekommen
2. G-CSF
- G-CSF darf bei Sichelzellpatienten nicht angewandt werden. Es führt
über die oft extreme Granulozytose zu einer ausgeprägten
Viskositäts-Erhöhung und dadurch nicht nur zu schweren Schmerzkrisen
sondern u. U. auch zu ZNS-Infarkten
3. Corticoide
Schmerzkrisen
Grundprinzipien
der Therapie:
- HYDRIERUNG
- ANALGETIKA
- THERAPIE AUSLÖSENDER FAKTOREN
- PRÄVENTION
1. Hydrierung
Wenn möglich (bei leichten Schmerzen, solange Darmgeräusche
vorhanden) oral bzw. über Magensonde.
Bei starken Schmerzen, fehlenden Darmgeräuschen intravenös.
Wieviel? Gesamtflüssigkeit (oral + IV) maximal 1 - 1
1/2 x Erhaltungsbedarf (bei pulmonaler Symptomatik bzw. Akutem
Thoraxsyndrom cave Überwässerung: nie mehr als Erhaltungsbedarf (= 1500
ml / m2 )geben)
Was? 5% Glukose + 50 mMol Na+/l + Erhaltungsbedarf
an KCl
Kontrollen: Elektrolyte, Gewicht täglich,
Ausscheidung
2 . Analgetika
Angst vor Drogenabhängigkeit und Zweifel an der Stärke der geklagten
Schmerzen dürfen nicht verhindern, daß ausreichend starke Analgetika,
z.B. Opiate, gegeben werden. Evtl. Drogenabhängigkeit bei
Sichelzellpatienten ist nicht Folge von Morphin-Gaben zur Analgesie,
sondern resultiert aus psychosozialen oder beruflichen
Schwierigkeiten.
Bei stationärer Aufnahme wegen einer Schmerzkrise soll angestrebt
werden, daß der Patienten spätestens nach Ablauf einer Stunde weitgehend
schmerzfrei ist. Da die meisten Patienten bereits ihre oralen Analgetika
(Stufe I + II) zu Hause genommen haben, ist bei stationärer Aufnahme
fast immer ein Opiat notwendig. Paracetamol oral ist nur wirksam bei
Kleinkindern und bei sehr leichten Schmerzen. Die altersabhängige
Höchstdosis muß beachtet werden.
Die Erfahrung, daß die Schmerzen beherrschbar sind, ist für den
Patienten ungemein wichtig und vermindert die Angst, die
schmerzpotenzierend ist. Bei allen Sichelzellpatienten
mit Schmerzkrisen, die einen Hb-Wert > 9 g/dl haben,
sollte ein Aderlass durchgeführt werden zur Senkung der
Viskosität
Prinzipien der Analgetikagabe in der Schmerzkrise
- DEM PATIENTEN DIE SCHMERZEN
GLAUBEN
- ausreichend starkes Mittel (keine
retard-Präparate!)
- ausreichend hoch dosieren
- ausreichend oft geben in festen
Intervallen
- ausreichend lange geben
- bei Besserung Reduktion der Einzeldosis,
nicht Änderung der Zeitintervalle
- nach mehrtägiger Morphiumgabe langsame
Dosisreduktion (evtl. mit Retardpräparat Oxygesic oder
MST wenn Patienten nach Hause gehen
wollen)
Die meisten Schmerzkrisen gehen mit
Temperaturanstieg und einer CRP-Erhöhung einher auf Grund
einer Interleukin-1-Ausschüttung. Patienten, die älter
sind als 5 Jahre, und die keine lokalen
Entzündungszeichen haben, brauchen bei Schmerzkrisen
mit Fieber und fehlendem klin. Hinweis auf Infektion
nicht antibiotisch behandelt werden.
Analgetika, die sich bei Schmerzkrisen bewährt
haben
A. leichte Schmerzen
|
Max. Dosis mg |
Applikation |
Intervall |
| 1. Paracetamol |
15 mg / kg / Dosis |
p.o. |
alle 4-6 h |
| 2. Novalgin |
15 mg / kg / Dosis |
p.o. |
alle 4-6 h |
| 3. Ibuprofen |
10 mg / kg / Dosis |
p.o. |
alle 8 h |
B. mäßig schwere Schmerzen
eines der unter A genannten Analgetika
plus
|
Max.
Dosis mg |
Applikation |
Intervall |
| Tramadol |
1 - 2 mg / kg / Dosis |
p.o. |
alle 4 - 6 h |
C. schwere Schmerzen
eines der unter A genannten Analgetika
plus
|
Max. Dosis
mg |
Applikation |
Intervall |
| Morphin |
0,1 - 0,15 mg / kg
/
Bolus-Dosis
evtl.
Dauerinfusion
Morphin
(0,03
mg/kg/h, b.B.
steigern auf 0,05 mg/kg/Std) oder
PCA
(Patienten-kontrollierte
Analgesie) |
i.v.
evtl. auch s.c.bei schlechten
Venen |
initialer Bolus,
Wiederholung nach 20
min. dann Dauerinfusion |
Unter IV-Gabe von Opiaten muß eine
Hypoventilation vermieden werden. Geeignete Maßnahmen
sind (neben der Überwachung der Oxygenierung und der
Vital-Parameter) ATEMGYMNASTIK bzw. Blähen der Lunge
mit SPIROMETER (z.B. Triflow) 10 Hübe alle 2-3 Stunden.
Bei Thoraxschmerzen und/oder sonstige pulmonaler
Symptomatik darf die Gesamt-Flüssigkeitsmenge den
Erhaltungsbedarf ( = 1,5 l / m2) nie übersteigen:
Gefahr des Lungenödems
Wenn Schmerzen abnehmen, parenterale Dosis um 10-20% reduzieren,
Zeit-Intervall aber beibehalten. Umsetzen auf orale Analgetika, wenn 50%
der initialen parenteralen Dosis erreicht ist. Morphin nicht abrupt
absetzen, sondern ausschleichen. Wenn Patienten sehr schnell die Klinik
verlassen wollen, noch einige Tage Oxygesic (Retard-Codein) oder MST
(orales Morphin-Retardpräparat). In der akuten Schmerzkrise sind
Retardpräparate kontraindiziert, da, wie der Name sagt, die Wirkung erst
nach 1-1,5 Tagen eintritt.
3. Therapie auslösender
Faktoren
Bei jeder Schmerzkrise, vor allem, wenn sie mit Fieber einhergeht,
muß eine Infektion (Abdomen, Harnwege, Lunge, Skelettsystem) gesucht und
dann gezielt behandelt werden.
4. Prävention
Schmerzkrisen können ausgelöst werden durch Ereignisse wie
Infektionen, Unterkühlung, Dehydrierung, Alkohol, aber auch durch
ungewöhnlich hohe Hb-Spiegel (bei HbSS / HbSß°Thal Patienten Hb > 10
g/dl, bei HbSC-Patienten Hb > 12-13 g/dl). Im Fall ungewöhnlich hoher
Hb-spiegel kann ein Aderlass erheblich zur Schmerzlinderung beitragen.
Patienten müssen informiert werden über ausreichende Flüssigkeitszufuhr
bei hohen Aussentemperaturen, Fieber, körperlicher Anstrengung mit
Schwitzen, Flugreisen (extrem trockenen Luft im Flugzeug!!) sowie über
Vermeiden von Unterkühlung und großen Alkoholmengen. Alle HbSS,
HbSD, HbSOArab bzw. HbSß°Thal Patienten sollen
Hydroxycarbamid bekommen zur Verhinderung von Schmerzkrisen.
Die Anfangsdosis bei Kindern ist 20 mg/kg/Tag, bei
Erwachsenen 15 mg/kg/Tag, Tagesmenge in einer täglichen
Dosis. Steigerung alle 6-8 Wochen ( wenn keine
Thrombopenie < 80 G/l bzw. keine Granulopenie < 2
G/l auftritt) bis zur Maximaldosis von 35 mg/kg/Tag.
Bei Erreichen der Zieldosis BB-Kontrolle alle 3 Monate.
Nebenwirkungen sind vor allem die Suppression der
Hämatopoese, mögliche Azoospermie bei postpubertären
männlichen Patienten. Hydroxycarbamid hat sich bei
einer maximalen Dosis von 35 mg/ kg KG nicht als
teratogen erwiesen (s. Kapitel Schwangerschaft). Es ist
vertretbar, es auch während der Schwangerschaft weiter
zu geben.
Corticoide können bei Sichelzellpatienten starke
Schmerzkrisen auslösen ( Viscositäts-Erhöhung durch die Granulozytose)
und sollten, wenn überhaupt, nur in lebensbedrohlichen Situationen
gegeben werden.
Aderläße
Patienten mit Sichelzellkrankheit SC haben häufig
relativ hohe Hb-Werte (10 - 13 g/dl). Vor einer Flugreise muß bei
Patienten, deren Hb höher als 11,5 g/dl ist, ein Aderlass durchgeführt
werden (10-15 ml /kg Körpergewicht Blut entfernen bei gleichzeitiger
Gabe von 0,9% NaCl in gleicher Menge) um den Hb-Spiegel < 10 g/dl zu
senken. Die hohe Viskosität des Blutes dieser Patienten kann sonst durch
die trockene Luft im Fluzeug so verstärkt werden, dass es zu schweren
Schmerzkrisen während oder nach dem Flug kommt.
Es gibt Hinweise dafür dass Vitamin D - Mangel die
Schmerz-Situation verschlechtert. Vor allem bei dunkelhäutigen Patienten
sollte nach einem Vit D Mangel gesucht und gegebenenfalls eine
Behandlung eingeleitet werden.
Crizanlizumab, einem Antikörper gegen P-Selectin (vermittelt Kontakt
der Thrombozyten,Granulozyten und Erythrozyten mit dem Endothel) wurde
wegen Wirkungslosigkeit im Vergleich zu Plazebo 2023 in Europa die
Zulassung entzogen.
Voxelotor wurde wegen Toxizität 2024 die Zulassung für Europa entzogen
5. Behandlung
schwerer,Morphin-resistenter Schmerzkrisen
Bei schweren Schmerzkrisen, die trotz ausreichender Opiatgabe nicht
beherrschbar sind, hat sich die Gabe von Ketamin
(Ketanest) bewährt. Dosierung: Beginn mit 2,0 µg / kg / min und
Steigerung bis zum Maximum von 3,0 µg / kg / min.
Sichelzellpatienten, vor allem Erwachsene, können neben nociceptiven
(die auf Opiate reagieren) auch neuropathische Schmerzen haben. Bei
diesen Patienten, die auf die üblichen Opiat-Dosen keine Erleichterung
ihrer chronischen Schmerzen haben, können Antiepileptika wie
Carbamazepin oder Gabapentin eingesetzt werden. Auch Cannabis-Derivate
sind bei neuropathischen Schmerzen wirksam
Schmerzkrisen bei Kleinkindern
Bei Kleinkindern ist die erste Manifestation der Sichelzellkrankheit
oft das sog. Hand - Fuß - Syndrom.
Durch Infarkte der Mittelhand-bzw. Fußknochen kommt es zu schmerzhaften
Schwellungen der Hände und Füße. Die Kinder wollen nicht mehr laufen
bzw. können die Hände nicht mehr benutzen zum Greifen. Meist ist ein
Basis-Analgeticum (z.B. Paracetamol) ausreichend zur Analgesie. Das
Hand-Fuß-Syndrom kommt bis zum 3. Lebensjahr vor. Danach sind
Schmerzkrisen dieser Lokalisation nicht mehr möglich, da die Hämatopoese
sich dann nicht mehr in den Mittelhand - bzw. Mittelfuß-Knochen
abspielt.
Osteomyelitis / Septische Arthritis
Wenn bei febrilen Patienten Knochen-bzw. Gelenkschmerzen und
Schwellung über den schmerzenden Partien mehr als 5 Tage anhalten, muß
eine Osteomyelitis / eitrige Arthritis ausgeschlossen werden: Blut-und
Stuhlkulturen (Salmonellen!), Ultraschall der betroffenen Stelle:
subperiostaler Erguss > 6 mm spricht für Osteomyelitis. Erguß
punktieren für Bakteriologie, evtl. Gelenkaspiration. Es gibt
kein bildgebendes oder nuklearmedizinisches Verfahren
das in der Lage ist, zwischen Infarkt und Osteomyelitis
zu differenzieren! MR bzw. Knochenszinti kann nicht zwischen
Infarkt und Osteomyelitis unterscheiden.
Differentialdiagnostisch sehr wichtig ist
die Schmerzqualität. Patienten mit Osteomyelitis
/Arthritis, die älter als 6 Jahre sind können angeben,
dass diese Schmerzen “anders” sind als bei
Schmerzkrisen.
Therapie mit Antibiotika, die wirksam sind gegen
Salmonellen, Staphylokokken, Streptokokken, H. influenzae.
Fieber
Alle Sichelzellpatienten, die anhaltendes Fieber >38,5°C haben
(zwei Messungen im Abstand von 1 Stunde) ohne klin. Zeichen einer
Infektion (Luftwege, GI, ) müssen untersucht werden. Auch erwachsene Sichelzellpatienten haben ein erhöhtes Sepsis-Risiko.
Sichelzellpatienten, die ambulant antibiotisch behandelt werden, müssen
täglich gesehen werden. Antipyretika dürfen, vor allem bei Kindern <
5 Jahren, auf keinen Fall gegeben werden, um das klinische Bild nicht zu
verschleiern.
Patienten < 5 Jahre:
- stationäre Aufnahme, unabhängig vom
klinischen Zustand, wenn bei der Untersuchung nicht
eine eindeutige Fieberursache gefunden wird: HWI,
Infekt d. ob. Luftwege, Gastroenteritis
Patienten > 5 Jahre:
- stationäre Aufnahme abhängig vom klinischen
Zustand
- wenn ambulant, tägliche
Kontrolle
Untersuchungen:
Labor:
- Blutbild, Retis, Blutkultur
je nach Klinik:
- Röntgen Thorax
- Urinstatus, Urinkultur
- evtl. Rachenabstrich
- evtl. Mycoplasmentiter
- LP bei Meningismus
bei Vd. auf Osteomyelitis:
- lokales Röntgen, Ultraschall,
- Aspirat von Ergußsubperiostal bzw. Gelenk,
- Stuhlkultur (Salmonellen?)
Antibiotika: müssen wirksam sein gegen S.pneumoniae
und H.influenzae und evtl. gegen Salmonellen ( Ciprofloxacin)
Bei Fieber unklarer Ursache: alle Kinder
< 5 Jahre, auch bei gutem AZ, und alle Patienten
> 5 Jahre die krank wirken
- Ampicillin oder Cefotaxim I.V.
Bei Meningitis vor
Erreger-Identifizierung:
- Cefotaxim ( CAVE: nach Gabe von
Ceftriaxon wurde bei Sichelzellpatienten letal
verlaufende Hämolysen beobachtet!)
Bei Verdacht auf Osteomyelitis:
- Ampicillin + Oxacillin bzw. Clindamycin
- bei Verdacht auf Salmonellen-Osteomyelitis: Ciprofloxacin
Bei abdomineller Symptomatik:
- Ampicillin + Metronidazol
Cave:Salmonellensepsis bei
Sichelzellpatienten geht
mit einer Mortalität
von ca. 25% einher. Häufig Multiorganversagen.
! Frühzeitige Austauschtransfusion indiziert !
Transfusionen/Aderlässe
Transfusionen bzw. partielle Austauschtransfusionen sind bei
Sichelzellpatienten nur in aussergewöhnlichen Situationen indiziert, um
entweder den akut abgefallenen Hämoglobinspiegel zu heben oder akut oder
über längere Zeit den Anteil an HbS zu senken. Indikationen und Art der
Transfusion sind aus der Tabelle 1 ersichtlich. Bei Individuen mit
Sichelzell-ßThalassämie besteht keinerlei Indikation für ein chronisches
Transfusionsprogramm wie bei der Thalassämia major, da es bei dieser
doppelt heterozygoten Erkrankung nicht zu einer klinisch relevanten
ineffektiven Erythropoese kommt.
Hauptindikationen für
Transfusionen bzw. partielle
Austauschtransfusion
Art der
Transfusion |
Indikationen:
Transfusion
meistens
notwendig |
Indikationen:
Transfusion
manchmal
notwendig |
| einmalige T. |
- Aplastische Krise
- Milzsequestration
- Akutes
Thorax-Syndrom |
- Lebersequestration
- Blutung (Hämaturie,
Gastrointestinal
Trakt, Gynäkologie)
- gesteigerte Hämolyse |
| Austausch-T. |
- ZNS-Infarkt/Blutung
- schwere
Infektionen
-
Multiorganversagen
- vor neurochirurg. und
ophthalmol.
Eingriffen |
- Thorax-Syndrom
- Girdle-Syndrom |
| chronische T. |
- nach ZNS-Ereignis
- chronische renale,
pulm. oder kardiale
Probleme |
Schmerzkrisen trotz Hydroxyurea |
Technik des manuellen partiellen
Austausches:
75% des Blutvolumens des Patienten ( d. h. 75 ml / kg KG x 0,75) wird
entfernt und mit der gleichen Menge Ery-Konzentrat + physiologische Na
Cl ( im Verhältnis 2 : 1) ersetzt. Beispiel: für den partiellen
Austausch eines 30 kg schweren Patienten müssen 1700 ml Blut entfernt
und mit 1130 ml Ery-Konzentrat + 570 ml physiologischer Kochsalzlösung
ersetzt werden. Es ist nicht notwendig, FFP zu verwenden. Der Austausch
kann entweder über einen zentralen Katheter oder über zwei großlumige
periphere Katheter, einer in jeder Ellenbeuge, durchgeführt werden. Die
Dauer des Austausches sollte 2 ½ Stunden nicht überschreiten. Um eine
HbS-Konzentration von < 30% zu erreichen, kann es notwendig sein, den
partiellen Austausch am nächsten Tag zu wiederholen.
Optimal ist eine Austauschtransfusion mittels Erythrozytapherese, da
es dadurch, wenn korrekt durchgeführt, nicht zu einer Eisenüberladung
kommt .
Technik des “modifizierten partiellen
Austausches”, z. B. bei Akutem Thoraxsyndrom, um
Transfusion effektiver zu machen, vor allem bei
Ausgangs-Hämoglobin > 6 g/dl:
- Aderlass von 10-15 ml/ kg KG des Patienten, über 2. Zugang die
gleiche Menge 0.9% Na Cl; anschließend Transfusion von EK
Vorsichtsmaßnahmen bei chronischem
Transfusionsprogramm:
- Untergruppengleiches Blut (Rhesus, Kell)
Verzögerte Hämolytische
Transfusions-Reaktion (DHTR) bzw. Hyper -Hämolyse-Syndrom
(HHS). Eine seltene aber lebensbedrohliche
Transfusions-Komplikation, die überwiegend bei Patienten mit
Hämoglobin-Krankheiten auftritt, die transfundiert wurden bei
Schmerzkrisen oder Akutem Thoraxsyndrom. Bei DHTR kommt es zur
Zerstörung der transfundierten, bei HHS sowohl der transfundierten als
auch der eigenen Erythrozyten durch Allo-und Auto-Antikörper sowie eine
Complement-Reaktion. Klinisch manifestiert es sich 6 - 10 Tage nach
einer Transfusion durch Schmerzen wie bei einer Schmerzkrise, Fieber und
einem drastischen Hb-Abfall, Hämoglobinurie, Retikulopenie (bei HHS),
Anstieg der LDH. der Coombs-Test kann, muß aber nicht positiv sein. Es
darf auf keinen Fall transfundiert werden. Therapeutisch sind eingesetzt
worden Corticoide, IVIG, Rituximab, Epo und Eculizumab. (s. Literatur
Pirenne)
Cave Hyperviskositäts-Syndrom: Es muß
dringend davor gewarnt werden, Sichel
zellpatienten, die keine akuten Probleme haben, über den für sie “normalen” Hb-Wert
(üblicherweise zwischen 6 - 8 g/dl) zu transfundieren lediglich in der
Absicht, ihr Hämoglobin in einen “normalen” Bereich zu bringen (Serjeant
2003).Durch Gefäßreaktionen kommt es zum sog. Hyperviscositäts-Syndrom,
das gekennzeichnet ist durch arteriellen Hypertonus, Krampfanfälle, Hirnblutung und evtl. Tod
Chelat-Therapie
Dauertransfundierte Sichelzellpatienten entwickeln auf Dauer eine
Eisenüberladung. Das Serum-Ferritin ist bei diesen Patienten nicht
aussagekräftig, da es beeinflußt wird u.a. von Schmerzkrisen,
Infektionen und Inflammation. Indikation zur Chelattherapie ist das
Ausmaß der Eisenbelastung der Leber, die gemessen wird durch eine
MR-Methode, das sog. Ferriscan. Ferriscan-Untersuchungen sind an 19
Kliniken bzw. Praxen in Deutschland möglich. Auskunft erteilt
http://www.resonancehealth.com
An einigen Kliniken gibt es MRT - Methoden der Lebereisenmeßung, die
mit Ferriscan vergleichbare Ergebnisse liefern.
Aderlässe
Aderlässe sind bei Sichelzellpatienten notwendig in Situationen, in
denen die hohe Viskosität des Blutes
zu Krankheitsmanifestationen führt. Dies ist relativ häufig der Fall bei
HbSC-Patienten, deren Hämoglobinwerte 13 g/dl erreichen können.
Bei Patienten mit Hörsturz muß sofort ein
Aderlaß durchgeführt werden, da der Hörverlust sonst
irreversibel ist. Führt der Aderlaß nicht zur Besserung
muß vor Cortisongabe eine Austauschtransfusion durchgeführt werden. Bei
HbSS bzw. HbSßThal Patienten steigt unter der Behandlung mit
Hydroxycarbamid nicht nur das HbF sondern auch das Gesamt-Hb an.
Bei Hb-Werten > 9 g/dl können vermehrt Schmerzkrisen
auftreten, aber auch Schwindelattacken oder
Hörstürze. In einer solchen Situation sollte durch Aderlässe
ein Hb-Wert < 9 erzielt werden. Bei HbSC-Patienten mit häufigen
Schmerzkrisen, die ein Hb > 10 haben, ist Hydroxycarbamid absolut
kontraindiziert, da es nicht nur den HbF-Anteil im Blut vermehrt,
sondern auch zu einem Anstieg des Gesamt-Hämoglobins führt. Diese HbSC
brauchen regelmäßige Aderlässe.
Technik des Aderlasses: Entnahme von 10 - 15 ml Blut / kg Kg und
gleichzeitig oder anschließende Gabe der gleichen Menge 0,9% Na Cl. Eine
entsprechende Flüssigkeitsmenge kann auch getrunken werden.
HbSC - Patienten, deren Hb > 11,5 g/dl ist, brauchen vor einem
längeren Flug ( > 6 Std.) einen Aderlass um Schmerzkrisen während
oder nach dem Flug zu verhindern.
Aderlässe können nicht die Entstehung der
proliferativen Retinopathie bei HbSC-Patienten verhindern, können aber
oft ein Fortschreiten der retinalen Veränderungen aufhalten.
Milzsequestration
Vorkommen
- bei HbSS-Patienten: meist Kinder < 6 Jahre
- bei Patienten mit doppelt heterozygoten Sichelzellkrankheiten
(Sichelzell-ßThalassämie, HbSC-HbSLepore - Erkrankung) möglich bis ins
Erwachsenenalter
- oft ausgelöst durch Infektion (Pneumokokken; Parvovirus B19!!)
Klinik und Labor
- Milz sehr groß, manchmal druckschmerzhaft
- Hypovolämischer Schock
- Hb-Abfall >3g/dl unter Normalwert für den Patienten (= große
Milzsequestration)
- Hb-Abfall <3g/dl unter Normalwert für den Patienten (= kleine
Milzsequestration)
- ausgeprägte Retikulozytose (oft 400-500%0), evtl. Thrombopenie; bei
gleichzeitiger Parvovirus B19 Infektion Retikulopenie!
Therapie
1. akut
- Volumen auffüllen
- sofortige Transfusion (Hb von 8g/dl anstreben; nach Gabe von 50% des
berechneten Transfusionsvolumens Pause einlegen, Hb bestimmen; restliche
50% des Blutes nur geben, wenn Hb von 8 g/dl noch nicht erreicht)
- sorgfältige Überwachung des Kreislaufes, am besten durch Messen des
zentralen Venendruckes, da unter Transfusion das in der
Milz gepoolte Blut wieder mobilisiert wird.
Cave Hyperviskositäts-Syndrom!
- Penizillin I.V. (nach Abnahme von Blutkulturen)
2. nach akuter Episode
Mehr als die Hälfte der Kinder haben erneute Milzsequestrationen
innerhalb kurzer Zeit. Das chronische Transfusionsprogramm bis zum
Erreichen des 2. Lebensjahres, das vor einigen Jahren empfohlen wurde,
hat sich nicht bewährt.
Prophylaxe
Eltern von Säuglingen und Kleinkindern mit Sichelzellkrankheit müssen
das Palpieren der Milz lernen, bei jedem Wickeln anwenden und bei
Tastbefund unter li Rippenbogen die Klinik aufsuchen.
Leber-Sequestration
Vorkommen
- alle Altersgruppen, meist bei Infekten
Klinik und Labor
- vergrößertes Abdomen, epigastrische Schmerzen
- vergrößerte, schmerzhafte Leber, gelegentlich Ikterus (direktes Bili
erhöht, Transaminasen evtl. leicht erhöht)
Therapie
- I.V.Hydrierung, Analgetika, Antibiotika; evtl. partielle
Austaustransfusion
Hepatopathie, chronisch
Vorkommen
- je älter Sichelzellpatienten werden, desto häufiger treten
Leber-Schädigungen auf (Patienten ab 25. - 30. Lebensjahr), die, wenn
nicht erkannt bzw. nicht behandelt werden, zur Zirrhose führen
Ursachen
- virale Hepatitiden
- Autoimmun-Hepatitits
- Eisenüberladung durch Transfusionen ohne ausreichende
Chelat-Therapie
- Alkoholabusus (selten bei Sichelzellpatienten)
- Sichel - Hepatopathie durch rezidivierende
intrahepatische Vaso-Okklusionen
Klinik und Labor
frühes asymptomatisches Stadium:
erhöhtes direktes Bilirubin,erhöhte gamma gT,
späteres Stadium: hohes direktes Bili, erhöhte gamma GT und Transaminasen,
Hypo-Albuminämie
spätes Stadium: zusätzlich pathol. Fibroscan, sehr häufig auch Proteinurie
und Niereninsuffizienz (Hepato-renales Syndrom)
Zirrhose
mit Ascites, Ösophagus-Varizen
Therapie
bei Eisenüberladung effiziente Chelat-Therapie, Dokumentation der
Leber-Eisenüberladung durch FerriScan
wenn hohes direktes Bilirubin nicht durch Steine in den
intrahepatischen Gallenwegen erklärt werden kann, Nachweis der
Leberschädigung durch Biopsie und Etablierung eines chronischen
Austauschtransfusions-Regimes (wenn möglich Erythrozytapherese); damit
kann die Entwicklung zur Zirrhose verhindert werden
Aplastische Krise
Vorkommen
- möglich bei allen Patienten mit chronischer hämolytischer Anämie,
d. h. auch bei Sichelzellpatienten
Ursache
- Parvovirus B19; selten andere Erythro-Viren
Klinik
- fieberhafter Infekt, oft schweres Krankheitsgefühl,
Nackensteifigkeit, Kopfschmerzen; in ca. 20 % wird Milzsequestration
ausgelöst!
kein Exanthem; Patienten sind infektiös
während der Aplasie!
Diagnose
- innerhalb von wenigen Tagen Abfall des Hb auf 3-4g/dl; völliges
Fehlen der Retikulozyten;
- Parvovirus-IgM-Titer positiv
Therapie
Verlauf
- Aplasie dauert meist 5-7 Tage
- Parvovirusinfektion hinterläßt lebenslange Immunität
Akutes Thorax-Syndrom
Thorax-Syndrom (ATS)
Ursache
in ca. 40-50% Fettembolien aus dem Knochenmark (Schmerzkrise
vorher!)
nach operativen Eingriffen, wenn nicht auf Vermeiden von
Hypoventilation geachtet wird
Hypoventilation bei schlecht überwachter Analgesie mit hohen
Opiat-Dosen
Überwässerung bei Behandlung einer Schmerzkrise
seltenere Ursachen: Infekte (viral, Mycoplasmen, sehr selten
bakteriell), Lungenödem durch Überwässerung
Alle diese auslösenden Ursachen können zu einer Gefäßreaktion führen,
die vergleichbar ist mit einer Sequestrationskrise in anderen Organen:
beim klassischen ATS sinken Hb und Thrombozyten
rapide ab.
Vorkommen
Klinik und Diagnostik
- sehr häufig vorausgegangene Schmerzkrise
- Thoraxschmerzen mit T-shirt-Ausbreitung
- Fieber, Tachypnoe, Tachycardie
- Husten ist spätes Symptom
- Röntgen: beidseits basal beginnende Konsolidierung
- Schmerzen, Tachypnoe, Fieber, Tachycardie gehen röntgenolog. Zeichen
voraus!
Therapie
- Oxymeter-Kontrolle, bei Hypoxie O2 Gabe
- vorsichtige I.V. Hydrierung (cave Überwässerung!), Analgetika,
Antibiotika
- Spirometrie, Atemgymnastik
- bei bronchospasmus Inhalation von Bronchodilatoren
- frühzeitige Transfusion
- Hydroxycarbamid-Therapie nach erstem
ATS
Bei klinischer Verschlechterung oder PO2<
80 mmHg (<11KPa): Austauschtransfusion
Bei Azidose und/oder Hypoxie: Beatmung,
NO-Inhalation wird diskutiert
Es gibt keine Indikation zur Antikoagulation beim ATS
Girdle Syndrom
(Paralytischer Ileus)
Ursache
- wahrscheinlich Infarkte der Mesenterial-Gefäße
Vorkommen
Klinik und Diagnostik
- massiv geblähtes Abdomen, fehlende Darmgeräusche
- Erbrechen, Schmerzen um die Taille, keine Abwehrspannung
- Rö-Thorax: oft bds. basale Infiltrate
- Rö-Abdomen: maximal dilatierte Darmschlingen, Spiegelbildung
Therapie
- I.V. Hydrierung, keine orale Flüssigkeit
- Magensonde
- Analgetika, wenn andere Schmerzursachen (Appendizitis,
Cholezystitis, Ulcus) ausgeschlossen
- Austauschtransfusion bei Verschlechterung des Allgemeinzustandes und
Anhalten des Ileussymptomatik > 24 h
- Laparotomie kontraindiziert!! Chirurgen fernhalten!
Gallensteine
Vorkommen
- bei Patienten < 18 Jahre in 30 %
- bei Patienten > 30 Jahre in 70 %
Therapie
Cholezystektomie bei Beschwerden (Meteorismus, Völlegefühl,
Koliken); in einigen Zentren wird empfohlen, Gallensteine laparoskopisch
zu entfernen sobald sie diagnostiziert sind, auch ohne
Symptomatik
bei laparoskopischer Cholezystektomie weniger postoperative
Komplikationen
Hyperbilirubinämie-Syndrom
Vorkommen
Vor allem bei HbSS Patienten; bei Kindern: meist milder Verlauf, nur
hohes Bili, guter AZ; bei
Adoleszenten/Erwachsenen: meist schwerer Verlauf,
starkes Krankheitsgefühl, hohe Transaminasen, drohendes
Nierenversagen
Klinik
- extremer Ikterus mit (Erwachsene) oder ohne (Kinder) sonstige
Symptome bzw. Krankheitsgefühl
Diagnose
- Bilirubin > 13 mg/dl (oft 50 - 80 mg/dl), ca. 50% direkt;
Transaminasen normal bis stark erhöht, evtl. pathol. Gerinnungsstatus,
evtl. Nierenversagen
Therapie
- sofortige partielle
Austauschtransfusion wenn zusätzlich zur
Hyperbilirubinämie beeinträchtigter AZ, Transaminasenerhöhung, patho.
Gerinnung oder Nierenbeteiligung.
- wenn, wie meist bei Kindern, nur
Hyperbilirubinämie bei gutem AZ und normalen Transaminasen und
Kreatinin: Abwarten
Hypersplenismus
Vorkommen
Bei Patienten mit Sichelzell-ß+Thalassämie kann die Milz bis ins
Erwachsenenalter vergrößert sein. Sie hat bei diesen Individuen auch
noch eine Rest-Funktion. Ein Hypersplenismus mit Panzytopenie kann ab
dem 10. Lebensjahr entstehen
Viele chronisch transfundierte Patienten entwickeln
Hypersplenismus.
Bei Patienten, die Hydroxyurea nehmen, kann sich (bei jungen
Kindern) die Rückbildung der Milz verzögern bzw. (bei älteren Kindern
und Erwachsenen) die Milz wird wieder palpabel und es entwickelt sich
ein Hypersplenismus.
bei allen Individuen mit noch erhaltener Milz besteht das Risiko
der Milzsequestration, unabhängig vom Alter
Therapie
- Wenn die Splenomegalie mit extrem niedrigen Hb-Werten oder mit einer
Panzytopenie einhergeht, ist die Splenektomie
indiziert.
Hüftkopf- Humeruskopfnekrosen
Häufigkeit: Bis zum 30. Lebensjahr haben 30 - 50%
aller Sichelzellpatienten eine Aseptische Knochennekrose erlitten, meist
am Hüftkopf, seltener am Humeruskopf. Hüftkopfnekrosen können auch schon
vor dem 10. Lebensjahr vorkommen.
Risikofaktoren: HbSC, Sichel-ß Thal; sehr häufige
Schmerzkrisen; relativ hohes Hb und/oder niedriges MCV
Diagnostik: Röntgen bzw. MRT (Nekrosen sichtbar wenn
Rö noch unauffällig)
Frühsymptome: Schmerzen in der Leiste, im
Gesäß, im Knie bzw. Schmerzen beim Gehen
Therapie: Es gibt keine einheitlichen
Therapieempfehlungen für Sichelzellpatienten mit Hüftkopfnekrosen. Ziel
jeder Therapie muß sein, Schmerzen zu vermindern und die Funktion
möglichst lange zu erhalten. Folgende Verfahren werden angewendet:
Kinder < 10 Jahren:
- initiale kurze Ruhigstellung, Analgetika; dann wieder
Belastung;
- Entlastung durch Thomas-Schienen werden nicht mehr empfohlen, da sie
zu einer Muskelatrophie der ruhiggestellten Extremität führen
Patienten > 10 Jahren:
In frühen Stadien (Fikat Std. I - II, d.h. es ist noch nicht zu
Einbrüchen der Zirkumferenz des Gelenkkopfes gekommen) hat sich die
Schenkelhals-Bohrung (Core decompression, Styles 1996)) bewährt
(Entnahme eines Knochenzylinders aus dem Femurhals) zur Druckentlastung.
Die Injektion von autologen Stammzellen in den Bohrkanal wird empfohlen
(Hernigou 2008).
In späteren Stadien kann durch eine Umstellungs-Osteotomie
Schmerzfreiheit erreicht werden.
Die Endoprothese ist indiziert, wenn durch Anbohren
keine Schmerzfreiheit erzielt wird.
Je älter Sichelzellpatienten werden, desto häufiger treten auch
Humeruskopfnekrosen auf. Auch bei diesen Nekrosen ist
im Frühstadiumein Anbohren indiziert.
Es ist im Interesse unserer Patienten, die Betreuung einer so
komplexen orthopädischen Komplikation wie es die Hüftkopfnekrose
darstellt, in eine Hand zu geben.
Prof. Dr. M. Rudert an der Orthopädischen Universitätsklinik Würzburg
hat in den letzten Jahren Erfahrung gesammelt mit Sichelzellpatienten
und sich bereiterklärt, unseren Patienten mit Hüftkopf - bzw.
Humeruskopfnekrosen konsiliarisch zu Verfügung zu stehen bzw. sie zu
betreuen. Termine können vereinbart werden unter der Tel-Nr.
0931-8031102(Sekretariat)
Priapismus
Vorkommen
Männliche Patienten bzw. Eltern müssen
frühzeitig informiert werden über die Möglichkeit des
Auftretens eines Priapismus. Werden kurze,
rezidivierende Episoden von Priapismus (stuttering priapism)
dem betreuenden Arzt nicht mitgeteilt, werden sie nicht
behandelt. Das Risiko einer langandauernder
Priapismus-Episode ist dann groß.
Nächtlicher Priapismus scheint sehr häufig mit
nächtlichen Hypoxien einherzugehen. Deshalb wir heute
empfohlen, bei Priapismus im Schlaflabor nach
hypoxischen Episoden zu suchen. Wenn solche Episoden
auftreten, kann nächtliche Sauerstoffgabe Priapismus
verhindern.
Therapie bzw. Prophylaxe
1. rezidivierende, kurze Episoden (sog.
“stuttering priapism”)
Etilefrin (Effortil, alpha-adrenerger Agonist)
- 5 - 10 mg po abends alle 4 Std., Wecker stellen, um um 03.00 Uhr
eine Dosis zu nehmen. Dieses Regime 2 Wochen versuchen
wenn Episoden auch tags auftreten, Effortil 5 - 10 mg 2x über
Tag
Wenn kein Erfolg kann Cyproteron, Tadalafil bzw. Terbutalen
eingesetzt werden (Urologe!)
2. Priapismus > 3 Stunden
wenn keine Rückbildung unter Hydrierung, Wärme, Analgetika,
Blasenkatheter wird von einigen Arbeitsgruppen folgendes empfohlen:
Drainage der Corpora cavernosa über G 23 Butterfly, anschließend
1-2 x intracavernöse Injektion von 6 mg. Effortil.
alternativ (da Effortil zur Injektion seit 07/05 nur noch über
Internationale Apotheke erhältlich):
Methylenblau 25 mg (Kinder) bzw. 50 mg (Erwachsene) intracavernös
oder
Epinephrin (Suprarenin) 10 ml einer 1: 1 000 000 Lösung
intracavernös (Herstellung der Verdünnung: 1 ml einer 1: 1 000 Lösung
Epinephrin in 1 Liter 0,9% Na Cl)
bei rezidivierenden, langen
Episoden
- Patienten werden angelernt, selbst mit Butterfly-Nadel (G 25 oder
27) 6 mg Effortil intracavernös zu injizieren wenn unter Hydrierung
Priapismus > 1 Std. anhält
Nach partiellen Austauschtransfusionen bei Priapismus wurden
neurologische Komplikationen beschrieben.
Schmerzlose Hämaturie
Vorkommen
- bei ca. 4 % der HbS-Heterozygoten
- bei Homozygoten häufiger, vor allem in der Schwangerschaft
Ursache
- meist Papillen-Nekrose; in 80% Blutung aus linker Niere
Differentialdiagnose
- Glomerulonephritis, Tuberkulose, Steine, Tumor, Harnwegsinfekt
Diagnose
Therapie
Bettruhe, Hydrierung, Alkalisierung des Urins
Desmopressin IV oder Tranexamsäure 1 g alle 6 Std. po
evtl. partielle Austauschtransfusion bei Nichtsistieren der
Blutung
Proteinurie
Proteinurie kann das erste Anzeichen sein für eine sich entwickelnde
Niereninsuffizienz. Bei Patienten > 6 Jahren
jährlicher Urinstatus unerläßlich.
Wenn Proteinurie nachgewiesen, muß Proteinausscheidung im 24 Std-Urin
bestimmt werden. ACE- Hemmer werden inzwischen bei einer Proteinurie
> 0,3 g/l empfohlen. Dadurch kann in vielen Fällen eine
Niereninsuffizienz über Jahre hinausgeschoben werden. Die zusätzliche
Behandlung mit Hydroxycarbamid kann die Nierenfunktion günstig
beeinflussen.
Hyposthenurie
Vorkommen
- Bei fast allen Sichelzellpatienten ist spätestens ab dem 3.
Lebensjahr die Konzentrationsfähigkeit der Niere stark eingeschränkt.
Enuresis ist sehr häufig.
Therapie
Keine Flüssigkeitseinschränkung! Sichelzellpatienten sind zur
Verhinderung von Schmerzkrisen (Viskosität muß niedrig bleiben) auf
höhere Flüssigkeitszufuhr angewiesen als Gesunde. Die Flüssigkeitszufuhr
wird normalerweise durch das Durstgefühl geregelt.
Wichtig ist, Eltern und Kinder über die Zusammenhänge aufzuklären
und ihnen zu versichern, daß die Enuresis nur sehr selten bis ins
Erwachsenenalter bestehen bleibt.
Hyperurikämie
Fast alle Sichelzellpatienten > 10 Jahren haben erhöhte
Harnsäurewerte im Serum, die jedoch keiner Therapie bedürfen. Es kommt
extrem selten zu Gicht-Symptomatik.
ZNS-Infarkte
Vorkommen
- ohne Prävention 11% der HbSS, HbSß°thal, HbSD, HbSOArab - Patienten
bis zum 18. Lebensjahr
Klinik
fokale motorische Ausfälle, Hemiparese; bei Kindern mit
plötzlicher
Bewegungseinschränkung ohne Schmerzen(können nicht mehr stehen oder
laufen) muß an ZNS Infarkt gedacht werden
Bewußtseinstrübung, Sprach-oder Sehstörung
Diagnostik und Therapie
gründliche neurologische Untersuchung
sofortige partielle Austauschtransfusion um HbS auf < 30% zu
senken und das Ausmaß der zerebralen Nekrosen zu minimieren
NMR, wenn NMR nicht zur Verfügung steht, CT nach ca. 4
Tagen
wenn bei klinisch eindeutigem Bild MR bzw.CT negativ, muß ein MR-
Angio durchgeführt werden. Da in ca. 60% der ZNS-Infarkte mit Rezidiven
gerechnet werden muß, müssen Patienten nach ZNS-Infarkt lebenslänglich auf ein chronisches Austauschprogramm gesetzt werden, um das HbS <30% zu halten. Ein
durchgemachter ZNS-Infarkt ist eine Indikation zur
Stammzelltransplantation
Trans-Cranielle Doppler Sonographie = Methode zur Diagnose von
Gefäßstenosen. Nur die nach Adams standardisierte Methode ist
verläßlich.
Möglichkeit zur frühzeitigen Erfassung von Veränderungen der basalen
Arterien bei Patienten mit homozygoter Sichelzell- bzw. HbSD,
HbSOArabicum und HbSß0Thal-Krankheit im Alter von 2 - 16 Jahren.
Jährliche Messung, bei grenzwertigem Ergebnis Wiederholung nach 3
Monaten, bei pathol. Werten (> 200 cm/sec) Beginn eines chron.
Transfusionsprogrammes um das HbS < 30% zu halten. Nach 3 Monaten
TCDS Kontrolle und MRA. Wenn Normalisierung der Flußgeschwindigkeit und
kein Nachweis von Stenosen im MRA kann evtl. überlappend das
Transfusionsintervall verlängert werden unter gleichzeitiger
Hydroxycarbamid-Gabe. Bei Fortbestehen der pathol. Flußgeschwindigkeit
bzw. Vorhandensein von Stenosen muß das Transfusionsprogramm weiter
geführt werden. In diesem Fall besteht eine Indikation zur
Stammzell-Transplantation.
ZNS-Blutungen
Vorkommen
- meist ältere Kinder und Erwachsene zwischen 20 - 40 Jahren
Blutungen können sein: epidural, subdural, subarachnoidal,
intraventrikulär, intracerebral; keine Prävention möglich
Klinik
- oft treten vorher starke Kopfschmerzen auf, ohne fokale
neurologische Ausfälle
- Meningismus und Kopfschmerz bei subarachnoidaler Blutung
- sehr schlechte Prognose bei massiver Blutung
Diagnostik und Therapie
- CT
- partielle Austauschtransfusion
- Kraniotomie und Entleerung subduraler oder epiduraler Blutungen
- LP bei Verdacht auf subarachnoidale Blutung
- Angiographie und evtl. operative Entfernung von Aneurysmen bei
subarachnoidaler Blutung
- Dauer des notwendigen Transfusionsregimes unklar.
HNO Probleme
Hohe Blut-Viskosität kann zu vermehrten Schmerzkrisen, aber auch zu
Vaso-Okklusionen im Innenohr bzw. Gleichgewichtsorgan führen mit
Hörsturz bzw. Vertigo. Die Blut-Viskosität hängt ab vom
Hämoglobinspiegel bzw. Hct, aber auch von der Beschaffenheit der
Erythrozyten. Abnorm geformte Erythrozyten wie Sichelzellen oder
Targetzellen tragen erheblich zu den schlechten Fließeigenschaften des
Blutes bei diesen Patienten bei.
90% aller erwachsener Patienten mit der Sichelzellkrankheit HbSC
haben ein Hb > 10 g/dl (häufig sogar > 12 g/dl), d.h. die
Viskosität ist auf Grund der abnormen Form der Erythrozyten sehr hoch.
Auch HbSLepore Patienten brauchen Aderlässe, wenn das Hb ständig > 10
g/dl ist.
Ungewöhnlich hohe Hb-Werte (Hb > 9 g/dl) kommen aber auch bei HbSS
bzw HbSßThal Patienten vor, vor allem dann, wenn sie Hydroxycarbamid
(Litalir, Syrea, Siklos) nehmen.
1. Hörsturz
- bei Sichelzellpatienten mit Hörsturz muß der Hb-Spiegel bestimmt
werden. Bei einem Hb > 9 g/dl ist sofort ein Aderlaß
indiziert. Auf keinen Fall darf Cortison gegeben werden, bevor ein
Aderlass durchgeführt wurde, da sonst schwere Schmerzkrisen ausgelöst
werden. Durch den Aderlaß erübrigt sich in den meisten Fällen die
Cortison-Gabe. Sollte der Aderlaß nicht zur Verbesserung des Hörens
führen, muß, bevor Cortison gegeben wird, eine Austauschtransfusion
durchgeführt werden..
2. Schwindel
- bei Hb-Werten > 9 g / dl ist ein Aderlaß
indiziert. In manchen Fällen muß die Hydroxycarbamid-Dosis reduziert
werden.
Ophthalmologische Probleme
1. Proliferative Retinopathie
Vorkommen
- bei ca. 70% aller Patienten mit HbSC-Erkrankung (meist Patienten
westafrikanischer (bzw. Karibik + Süd-Amerika) Herkunft!) ab der 2.
Lebensdekade
- bei Patienten mit HbSS und Sichelzell-ßThalassämie wesentlich
seltener
Klinik
- Verschwommenes Sehen, Blindheit durch Glaskörperblutung bzw.
Netzhautablösung
Diagnostik
- ab dem 7. Lebensjahr muß die Retina von HbSC-Patienten, ab dem 10.
Lebensjahr die aller anderen Patienten mit Sichelzellkrankheit jährlich
bei völlig dilatierter Pupille untersucht werden.
Therapie
frühzeitige Laser-oder Photokoagulation der neovaskularisierten
Retinaareale
Einsatz von anti VEGF (vascular endothelial growth
factor)
chirurgische Therapie bei Glaskörperblutung bzw.
Netzhautablösung.
Vor ophthalmologischen Operationen immer partielle
Austauschtransfusion!
2. Traumatisches Hyphäma
Sichelzellpatienten mit traumatischem Hyphäma (Blut in der vorderen
Augenkammer) müssen sofort einem Ophthalmologen vorgestellt werden, da
sie ein hohes Risiko haben, gesteigerten Augeninnendruck zu
entwickeln
Unterschenkel-Ulzera
Vorkommen
Klinik
- offene, nässende, sehr schmerzhafte Ulzera, meist im
Knöchelbereich
Diagnostik
- Wundabstrich, systemische Antibiotika entsprechen dem Keim (meist
Staph aureus)
- bei chronischen Ulzera evtl. MRT um Osteomyelitis
auszuschließen
- auf keinen Fall: Biopsie des
Ulkus
Therapie: am Wichtigsten ist Zusammenarbeit
mit Wundexperten; wirksamste
Therapie-Modalität wäreRuhigstellung und Hochlagerung; aber kaum
durchführbar, da es sich um junge, aktive, z.T. berufstätige Patienten
handelt
Lokale Therapie: Sauberhalten;
Granulations-fördernde Verbände, evtl. Curettage von Belägen, Vermeiden
von Trauma; Kompressionsverbände
Hydroxycarbamid, falls nicht schon genommen;
Hydroxycarbamid löst bei Sichelzellpatienten entgegen einer früheren
Meinung, keine Ulzera aus.
auf keinen Fall: Spaltlappen-Deckung;
wird sofort abgestoßen; keine Transfusionen, um Hb
anzuheben
Schmerztherapie: ausreichend systemische
Analgetica der Stufe I (Novalgin) und Stufe II (Tramadol); evtl. Opiate
notwendig; lokal applizierte Morphin-Lösung: 0,1% Morphin - Lösung
oder in 1-2 ml aufgelöstes Oxycodon (5 oder 10 mg Tbl) können
systemische Opiate einsparen helfen
Einzelbeobachtungen: topisch appliziertes
GM-CSF; topisch appliziertes Gel aus Stammzellen + Thrombozyten;
topisches Na-Nitrat (2% Salbe); Fisch-Haut
Chirurgische Eingriffe
Folgendes Vorgehen wird heute empfohlen:
Partielle Austauschtransfusion
um das HbS auf < 30% zu senken:
1. bei schwerkranken, beeinträchtigten Patienten, die einen
unaufschiebbaren chirurgischen Eingriff benötigen
2. vor größeren Eingriffen wie Herz-oder Lungenoperation,
neurochirurgischen und ophthalmologischen Operationen, Eingriffen in
Blutleere.
Bei Patienten in gutem Allgemeinzustand ist
bei steady-state Hb - Werten um 7- 8 g/dl vor kleineren
Eingriffen (AT, TE, Circumcision) keine Transfusion
notwendig. Auch Splenektomie und Cholezystektomie, die
häufigsten operativen Eingriffe bei
Sichelzellpatienten, können bei Patienten in gutem AZ
ohne vorherige Transfusion durchgeführt
werden.
Bei jeder Allgemeinnarkose bei
Sichelzellpatienten ist auf folgendes zu
achten:
1. H y d r i e r u n g mit 1500 ml / m² 24 Std.
von Beginn der Nüchternheit bis zur vollen oralen Flüssigkeitszufuhr.
2. O x y g e n i e r u n g
von Prämedikation bis zum vollen Wachsein, unter
Oxymeterkontrolle.
3. V e r m e i d e n v o n U n t e r k ü h
l u n g
4. Postoperative Atemgymnastik
Bei größeren Eingriffen, vor allem bei Patienten mit
Sichelzell-ßThalassämie und HbSC-Erkrankung, wird empfohlen, bis zur
vollen Mobilisierung fraktioniertes Heparin s.c.zu geben.
Pubertät
ist verzögert, vor allem bei Knaben und
untergewichtigen Patienten; verzögerte Pubertät hängt mit verminderter
Muskelmasse bei Sichelzellpatienten zusammen.
Therapie
- Erklärung der Zusammenhänge Gewicht -und Pubertät und Versicherung,
daß Pubertät, wenn auch später, fast immer spontan eintritt.
- evtl. kalorienreiche Zusatznahrung in Form von hochkalorischer
Flüssignahrung
Kontrazeption
Risiken einer Schwangerschaft sind größer als Risiken der Pille
Es gibt keinen Nachweis, daß Sichelzellpatientinnen, die die Pille
nehmen, ein größeres Thromboserisiko haben als Gesunde.
Sichelzellpatientinnen, die um die Zeit der Menstruation gehäuft
Schmerzkrisen haben ( 30% aller Sichelzellpatientinnen!) profitieren von
einer überwiegend Progesteron-haltigen Pille.
Möglichkeiten der Kontrazeption:
1.Gestagen-Monopräparate
2. Depot-Präparat Medroxy-Progesteron I.M. alle 3 Monate( darunter
weniger Schmerzkrisen, aber Risiko der Osteoporose)
3. Intrauterine Spirale (kein erhöhtes Infektionsrisiko!)
Pränatale Diagnostik
Eltern, die schon ein Kind mit Sichelzellkrankheit haben und weitere
Kinder wünschen sowie Paare, die Träger der Sichelmutation bzw. der
ß-Thalassämie-Mutation sind, sollten frühzeitig über die Möglichkeit der
Pränatalen Diagnostik informiert werden.
Jedes Individuum hat ein Anrecht auf
Information über das genetische Risiko, ein krankes
Kind zu bekommen. Die persönliche Einstellung des
betreuenden Arztes zur pränatalen Diagnostik bzw. zum
Schwangerschaftsabbruch darf diese Informationspflicht
nicht beeinträchtigen. Die Entscheidung für oder gegen
pränatale Diagnostik ist eine Entscheidung der
betroffenen Eltern und nicht des Arztes. Es ist nicht
vertretbar bei Familien bestimmter
Religionszugehörigkeit (Islam, Katholizismus) von vorne
herein davon auszugehen, dass die Pränatale Diagnostik
sowieso nicht akzeptiert wird, es sich deshalb nicht
lohnt, überhaupt darüber zu sprechen. Auf Zypern
(griech. Orthodox, 19% ß-Thal Träger in der
Bevölkerung) und Sardinien (katholisch, 15% ß-Thal
Träger in der Bevölkerung) werden so gut wie keine
Kinder mit Thalassämia Major mehr geboren, dank
umfassendendem Träger-Screening und Information über
die Pränatale Diagnostik. Auch der Islam verbietet
den Schwangerschaftsabbruch aus medizinischen Gründen
nicht (siehe Literatur Zahed et al.)
Um festzustellen, ob der Fet homozygot HbSS bzw. doppelt heterozygot
(HbSD, HbSß Thal) oder gesund ist (HbAA, HbAS, HbAD oder ß Thal Träger)
werden in der 10. - 12. Schwangerschaftswoche durch Chorionzottenbiopsie
fetale Zellen gewonnen, die mit molekularbiologischen Methoden
untersucht werden. Ist der Fet betroffen, steht den Eltern die Option
des Schwangerschaftsabbruches offen. Ist der Fet lediglich Träger eines
der Merkmale (HbS, ß Thal) kann die Schwangerschaft ausgetragen werden
ohne Angst vor der Geburt eines kranken Kindes.
Die Pränatale Diagnostik für
Hämoglobinerkrankungen wird in Deutschland durchgeführt u.
a. bei
Institut für Humangenetik der Universität Münster, Vesaliusweg
12, 48129 Münster. Tel. 0251-8355432
Prof. Dr. A. Kulozik, Univ.-Kinderklinik Heidelberg, Im
Neuenheimer Feld 151, 69120 Heidelberg Tel 06221-564555
Dr. rer. nat. Christa Aulehla-Scholz, Institut für Klinische
Genetik, Olgahospital, Bismarckstr. 3, 70176 Stuttgart Tel 0711
278-74008
Pränatal Medizin München Lachnerstr. 20, 80639 München
Die Präimplantationsdiagnostik (PID) ist in Deutschland seit Juni
2012 erlaubt, es gibt aber kaum Erfahrung damit.Unsere Patienten haben sich bisher an Kliniken in Adana /Türkei und an die
Freie Universität Brüssel gewandt.
Schwangerschaft
Frauen mit Sichelzellkrankheit sind fertil, während die Fertilität
bei Männern durch geringe Spermienzahlen und herabgesetzte Motilität
vermindert ist.
Der Partner der Schwangeren muß so früh wie möglich untersucht
werden, ob er Träger einer Hämoglobinopathie ist, um pränatale
Diagnostik durchführen zu können.
Betreuung bei Sichelzellpatientinnen HbSS,
HbSßThal, HbSD, HbSOArab die Hydroxycarbamid (HU)
nehmen
Die offiziellen Empfehlungen sind immer noch, HU 3-6 Monate vor einer
geplanten Schwangerschaft abzusetzen. Die Wirklichkeit sieht anders aus,
da es unwahrscheinlich ist, dass eine Frau, die wieder von heftigen
Schmerzen geplagt wird, schwanger wird. Es ist vertretbar, das HU
entweder bei dokumentierter Schwangerschaft abzusetzen, aber auch, es
weiter zu geben. (Habibi Revue Med Interne 2015) Die hypothetische
teratogene Wirkung von Hydroxycarbamid (HU) beim Menschen beruht auf
Daten von Tierversuchen in den 60er und 80er Jahren, bei denen Ratten
bzw. Hamster ein Vielfaches (50-100x) der Dosis bekamen, die bei
Sichelzellpatienten angewandt wird . Bei Kindern, deren Mütter oder
Väter um den Zeitpunkt der Konzeption und/oder während der
Schwangerschaft HU genommen hatten, wurden keine Fehlbildungen gesehen
(Ballas 2009). Z.Z wird diskutiert, HU während der Schwangerschaft
weiter zu geben (Savage 2015).
Jede
Schwangerschaft einer Sichelzellpatientin ist eine
Hochrisiko-Schwangerschaft. Sorgfältige und regelmäßige pränatale
Betreuung ist wesentlich für den günstigen Verlauf der Schwangerschaft.
Sichelzellpatientinnen sollten doppelt so häufig gesehen werden
verglichen mit Schwangeren ohne Sichelzellkrankheit . Regelmäßige
Transfusionen während der Schwangerschaft vermindern die Morbidität der
Mutter, haben aber keinerlei positive
Auswirkung auf das Kind. Regelmäßige
Transfusionen sind indiziert bei Patientinnen, die in
der Vorgeschichte schwere Komplikationen hatten (Thoraxsyndrom,
ZNS-Infarkte, schwere Schmerzkrisen) bzw. bei denen Hydroxycarbamid
abgesetzt wurde und die jetzt wieder häufige/schwere Schmerzkrisen
haben.
Stillen ist erlaubt unter HU, auch von offizieller Seit (Ware 2020).
Ein Neubgeborenes einer Mutter, die die übliche Dosis HU einnimmt, müßte
10 Liter Muttermilch am Tag trinken, um eine pharmakologisch wirksame
Dosis HU zu bekommen.
Einzelne Transfusionen können indiziert sein bei
interkurrenten Komplikationen wie Akutem Thorax-Syndrom, symptomatischer
Anämie.
Bei Schmerzkrisen in der Schwangerschaft können folgende Analgetika
gegeben werden: PCM und Opiate während gesamter Schwangerschaft, NSAR
(z.B. Ibuprofen) nur bis zur 28. SSW. Novalgin darf in der
Schwangerschaft nicht genommen werden da es Hinweise gibt, dass es für
eine erhöhte Leukämie-rate bei den Kindern dieser Mütter verantwortlich
ist. Eine prompte Behandlung von Harnwegsinfekten ist wichtig.
Schwangere mit HbSC-Erkrankung oder HbSß+ Thal haben meist keine
wesentlichen Probleme und benötigen nur sehr selten Transfusionen.
Schwangere Sichelzellpatientinnen sollten frühzeitig (25. SSW)
Kontakt mit dem Zentrum, wo sie entbinden werden, aufnehmen.
Entbindung kann vaginal erfolgen. Die Indikation zur
Sectio wird auf Grund geburtshilflicher, nicht hämatologischer Kriterien
gestellt. Eine regionale Anästhesie wird empfohlen, auch für eine
Sectio.
Nach einer vaginalen Entbindung sollte 7 Tage lang, nach einer Sectio
6 Wochen lang Heparin s.c. gegeben werden, 2 x täglich 5000 I.U .
Knochenmarktransplantation-Stammzelltransplantation
Die Stammzelltransplantation (SZT) stellt eine Möglichkeit der
Heilung für Sichelzellpatienten dar.
Die Hauptschwierigkeit bei der Indikationsstellung ergibt sich aus
der Tatsache, daß es sich zwar um eine Erkrankung mit möglichen
lebensbedrohlichen Komplikationen handelt, daß aber der individuelle
Verlauf der Erkrankung nicht vorhersehbar ist.
Eine klare Indikation zur SZT ist: ein durchgemachter ZNS-Infarkt bei
einem Patienten bzw. auch alle Patienten auf chronischem
Transfusionsprogramm aus anderen Gründen (z.B. persistierende
pathologische Flußgeschwindigkeiten in der Transcraniellen
Doppler-Sonographie, schwere Schmerzkrisen bzw. gehäufte ATS bei
Patienten, die nicht profitieren von Hydroxycarbamid). Es wird
empfohlen, alle HbSS, HbSD, HbSOArab bzw. HbSß°Thal-Kinder, die einen
HLA-identischen Familienspender haben, zu transplantieren, auch ohne
vorherige klinische Probleme.
Inzwischen gibt es eine nonmyeloablative Konditionierung für
Erwachsene, die einen HLA-identischen Familienspender haben (Hsieh
2009).
Fremdspender - und haploidentische Transplantationen bei
Sichelzellpatienten sind noch als experimentell einzustufen und sollten
nur im Rahmen von Studien durchgeführt werden.
Wenn eine SZT in Betracht gezogen wird, sollte mit einem
Transplantationszentrum, das Erfahrung mit Sichelzellpatienten hat,
Kontakt aufgenommen werden, z.B. Prof. S. Corbacioglu Universitätsklinikum Regensburg
Seit Oktober 2019 läuft eine Studie, die Gentherapie für
Sichelzellpatienten anbietet. Es gibt damit noch sehr wenig
Erfahrung.
Hydroxycarbamid
Hydroxycarbamid, ein Zytostatikum, kann bei Sichelzellpatienten u. a.
über eine HbF-Synthesesteigerung und eine Veränderung der
Oberflächeneigenschaften der Erythrozyten die Häufigkeit und Schwere von
Schmerzkrisen und Akuten Thorax-Syndromen reduzieren. Hydroxycarbamid
wird inzwischen empfohlen für alle HbSS, HbSD, HbSOArab und HbSß°thal
Patienten ab dem 9. Lebensmonat, bei HbSß+Thal Patienten bei häufigen
schweren Schmerzkrisen, einem durchgemachten Akuten Thorax-Syndromen
bzw. bei ständiger schwerer Anämie (Hb < 6 g/dl). Es gibt inzwischen
Langzeit-Beobachtungen über > 25 Jahre. Teratogenität bei
therapeutischen Dosen wurde bis jetzt nicht nachgewiesen. Bis jetzt
wurde auch keine erhöhte Malignomrate bei behandelten Patienten
beobachtet (Zeitraum 17 Jahre). Die wichtigsten Nebenwirkungen sind:
Immunsuppression (opportunistische Infektionen), Azoospermie ,
Hypomagnesämie, Haut-und Nagel-Hyperpigmentierung, Gewichtszunahme,
Myelosuppression
HbSC-Patienten mit Hb-Werten > 10g/dl sollten kein Hydroxycarbamid
bekommen, sie profitieren von Aderlässen. Nur ca. 10% der erwachsenen
HbSC-Patienten haben ständig ein Hb< 10g/dl. Bei Schmerzkrisen kann
ein Versuch mit Hydroxycarbamid gemacht werden, u.U. begleitet von
Aderlässen.
Voraussetzung für eine Therapie mit Hydroxycarbamid ist Kontinuität
der ärztlichen Betreuung, regelmäßige Laborkontrollen (Blutbild und
Retis) zu erwartende Kompliance des Patienten bezüglich der häufigen
Laborkontrollen und Dokumentation von Nebenwirkungen. Jungen Männern
kann vor Beginn einer Hydroxycarbamid-Therapie eine
Sperma-Kryokonservierung angeboten werden. Die Kosten der
Kryokonservierung werden allerdings von den Kassen nicht übernommen.
Probleme erwachsener Sichelzellpatienten
siehe auch Leitlinien http://www.dgho.de/onkopedia/sichelzellkrankheit
Mit zunehmendem Alter leiden Sichelzellpatienten immer mehr unter
chronischen Organschäden.
Kardial:
- Myocardiopathie (durch Minderdurchblutung des Myocards)
Diagnose:
- Doppler-Echographie, Herzkatheter
Therapie: - chron. Transfusionsprogramm
Pulmonal:
- Lungenfibrose, Pulmonaler Hochdruck (u. a. Folge der intravasalen
Hämolyse)
Renal: oberer Normwert für Kreatinin ist bei
Sichelzellpatienten mit den schwer verlaufenden Formen
HbSS,HbSD, HbSOArab, HbSß°Thal 0,8 mg/dl.
- Niereninsuffizienz bei 5 - 10 % aller Patienten (Alter meist > 40
Jahre)
Diagnose:
ansteigendes Kreatinin (bei Kreatinin von > 0,8 mg/dl Verdacht
auf chron. Niereninsuffizienz!! Sichelzellpatienten haben normalerweise
sehr niedrige Kreatinin-Werte)
Zunehmende Anämie
Zunehmende Proteinurie trotz ACE-Hemmern
Therapie: - chron. Transfusionsprogramm - Dialyse;
evtl. Transplantation, dann allerdings gefolgt von chron.
Transfusionsprogramm
Hämatologisch:
- Knochenmarksinsuffizienz (Panzytopenie) bei Patienten > 40
Jahre
Ursache: ausgedehnte KM-Nekrosen
ZNS:
- mit zunehmendem Alter haben manche Patienten abnorme MRTs durch sog.
“silent infarcts”, die sich manifestieren können durch
Gedächtnisverlust, Kopfschmerzen, neuro-psychologische
Auffälligkeit
- ZNS-Blutungen
Hepatisch:
Leber-Infarkte können zu postnekrotischer Zirrhose
führen
Leber-Sequestrationen (Hepatomegalie, Hb-Abfall, Fieber,
eingeschränkte Leberfunktionen, erhöhtes direktes Bilirubin, schweres
Krankheitsbild).
Akutes Leberversagen bei
Hyperbilirubinämie; meist gefolgt von
Niereninsuffizienz und
Multiorganversagen
Hepatopathie mit Fortschreiten zur
Zirrhose; diagnostisch richtungsweisend sind
ansteigende Werte für das direkte Bilirubin
- Eisenüberladung durch Transfusionen ohne ausreichende
Chelierung
- Virus-Hepatitis
- Auto-Immun-Hepatitis
- Sichel-Hepatopathie (ohne die genannten Ursachen) durch
intrahepatische Gefäßobstruktion
Therapie: - Austauschtransfusion bei
Leber-Sequestration bzw. Akutem Leberversagen
- chronisches Austauschtransfusions-Programm (Erythrozytapherese) bei
beginnender Hepatopathie
Knochen:
- Deckplatteneinbrüche, Osteopenie, Asept. Knochennekrosen
Augen:
- Proliferative Retinopathie (vor allem bei HbSC-Patienten)
iatrogen:
Chronische Schmerzen
Erwachsene Sichelzellpatienten haben weniger häufig akute
Schmerzkrisen. Dafür können bei einigen älteren Patienten chronische
Schmerzen auftreten, die Patienten und Ärzte verunsichern:
- Patienten sind verunsichert, weil das Schmerzmuster plötzlich völlig
anders ist. Die mühsam gelernte Taktik, mit akuten Schmerzen, die
irgendwann wieder aufhören, umzugehen, funktioniert nicht mehr.
- Ärzte sind verunsichert, weil sie meisten nicht wissen, dass auch
chronische Schmerzen zum Krankheitsbild gehören können.
Sowohl Ärzte als auch Sichelzellpatienten
müssen akzeptieren, dass es im Rahmen der
Grunderkrankung zu chronischen Schmerzen kommen kann.
Den Patienten muß versichert werden, dass sie jetzt
nicht noch eine andere Erkrankung haben. Ausnahme:
gleichzeitige Rheumatoide Arthritis bei Patienten, die
Schmerzen in den kleinen Gelenken (Morgensteifigkeit
der Hände und Schmerzen in kleinen Gelenken!)
haben
Ursachen chronischer Schmerzen können
sein:
- Avaskuläre Nekrosen (Hüftkopf, Humeruskopf)
- Deckplatteneinbrüche der Wirbelkörper
- Arthropathien (vor allem Knie)
- Z. N. Multiplen Knocheninfarkten
Lediglich Schmerzen durch Avaskuläre Nekrosen können ursächlich
angegangen werden (s. S. 10).
In allen anderen Fällen muß durch Einsatz von Analgetika und
physikalischen Methoden versucht werden, die Lebensqualität des
Patienten zu erhalten. Bei Rückenschmerzen durch Deckplatteneinbrüchen
sollte auf keinen Fall ein Korsett getragen werden, das zur Atrophie der
Rückenmuskulatur führt. Wesentlich sinnvoller ist, zusätzlich zu
Analgetika, intensive Krankengymnastik zur Kräftigung der
Rückenmuskulatur.
Bewährt haben sich als Basisanalgetika die
Nicht-Steroidalen-Anti-Rheumatika. bei längerem Gebrauch muß allerdings
alle 3 Monate die Nierenfunktion überprüft werden. Wenn die NSAR nicht
ausreichen, müssen sie kombiniert werden mit Retard-Opiaten wie Oxycodon
(Oxygesic) oder Morphin (MST). Patienten müssen lernen, die
kleinstmögliche Menge zu nehmen, die ihre Schmerzen kontrolliert.
In manchen Fällen ist eine Änderung der Lebensgewohnheiten bzw. eine
Veränderung des Arbeitsplatzes (Umschulung) notwendig. Physiotherapie,
Wärme oder Akupunktur können hilfreich sein.
LITERATUR
Adams RJ, McKie VC, Hsu L et al: Prevention of first stroke by
transfusions in children with sickle cell anemia and abnormal results on
trancranial doppler ultrasonography. N Engl J Med 1998;
339:5-11
Hernigou P, Poignard A, Zilber S, Rouard H: Cell therapy of hip
osteonecrosis with autologous bone marrow grafting. Indian J Orthop
2008; 43: 40-45
.
Hsieh MM, Kang FM, Fitzhugh CD et al: Allogeneic hematopoietic
stem-cell transplantation for sickle cell disease. N Engl J med 2009;
361: 2309-2317
Serjeant G: Blood transfusion in sickle cell disease: a
cautionary tale. Lancet 2003;361:1659-1660
Styles LA, Vichinsky EP: Core decompression in avascular necrosis
of the hip in sickle-cell disease. Am J Hematol. 1996;
52:103-107
Zahed L, Bou-Dames J: Acceptance of first trimester prenatal
diagnosis for the haemoglobinopathies in Lebanon. Prenat Diagn 1997;
17:423-428
Weitere Literatur unter www.sichelzellkrankheit.de